 Back
BackReactions at the α-Carbon of Carbonyl Compounds: Mechanisms and Applications
Study Guide - Smart Notes
 Tailored notes based on your materials, expanded with key definitions, examples, and context.
Tailored notes based on your materials, expanded with key definitions, examples, and context.
Reactions at the α-Carbon of Carbonyl Compounds
Introduction to α-Carbon Reactivity
Carbonyl compounds are well-known for their reactivity at the carbonyl carbon, but many also exhibit significant reactivity at the α-carbon—the carbon adjacent to the carbonyl group. The hydrogen atoms attached to this α-carbon (α-hydrogens) are more acidic than typical sp3 C–H bonds, enabling a variety of important organic transformations.
The Acidity of α-Hydrogens
Factors Affecting Acidity
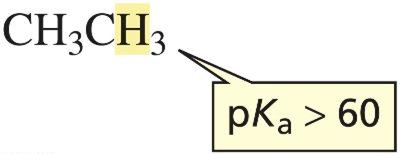
Electronegativity and Bonding: Hydrogens attached to sp3 carbons are usually not acidic due to similar electronegativities of C and H, resulting in high pKa values (e.g., ethane pKa > 60).
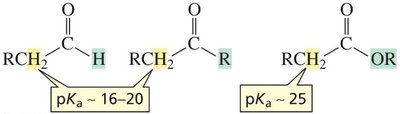
α-Hydrogens: The α-hydrogen of aldehydes and ketones has a much lower pKa (16–20), and esters have pKa ≈ 25, making them significantly more acidic than other sp3 C–H bonds.
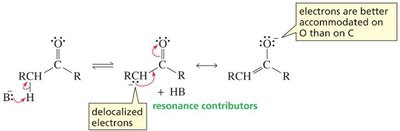
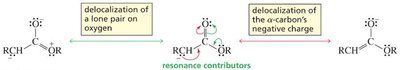
Delocalization: The increased acidity is due to the stability of the conjugate base (enolate ion), where the negative charge is delocalized onto the more electronegative oxygen atom.
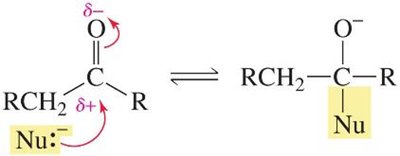
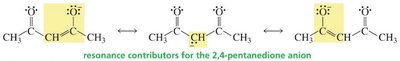
Resonance Stabilization of Enolates
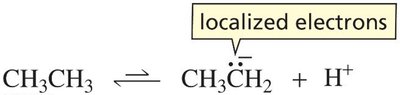
When a proton is removed from a simple alkane, the resulting carbanion is unstable due to localized electrons on carbon.
In contrast, removal of an α-hydrogen from a carbonyl compound forms an enolate ion, stabilized by resonance delocalization onto oxygen.
Comparing Aldehydes, Ketones, and Esters
Aldehydes and ketones are more acidic than esters because the lone pair on the ester oxygen competes with delocalization of the negative charge, reducing resonance stabilization.
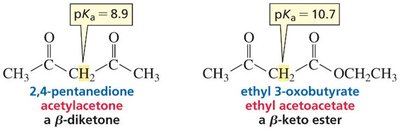
When the α-carbon is flanked by two carbonyl groups (e.g., β-diketones, β-keto esters), the acidity increases further due to additional resonance stabilization.
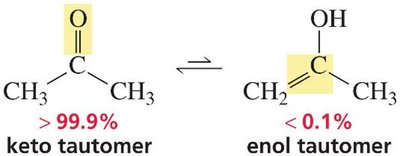
Keto-Enol Tautomerism
Definition and Equilibrium
Keto-enol tautomerism is the equilibrium between a ketone (or aldehyde) and its enol form. Tautomers are isomers that differ in the position of a hydrogen and a double bond. For most simple ketones, the keto form is strongly favored.
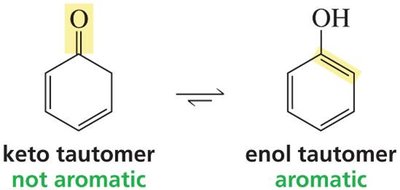
Phenol is an exception, where the enol form is stabilized by aromaticity.
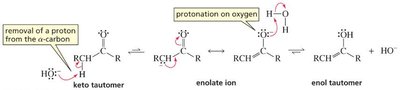
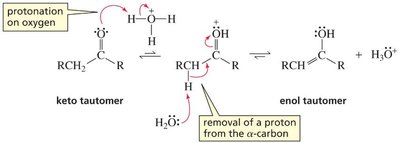
Mechanisms of Keto-Enol Interconversion
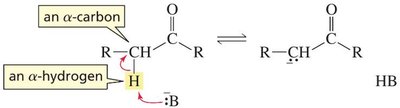
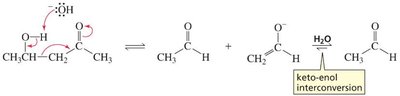
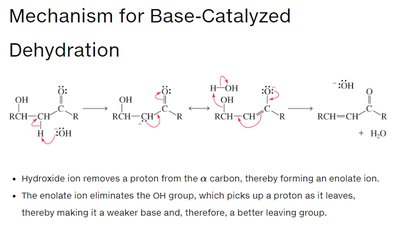
Base-Catalyzed: Hydroxide removes an α-hydrogen, forming an enolate ion, which is then protonated on oxygen to yield the enol.
Acid-Catalyzed: The carbonyl oxygen is protonated first, then water removes an α-hydrogen to form the enol.
The order of steps is reversed in acid and base catalysis, but both regenerate the catalyst.
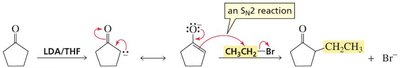
Alkylation of Enolate Ions
Formation of Carbon-Carbon Bonds
Alkylation of the α-carbon is a key method for forming new C–C bonds. The reaction proceeds via deprotonation of the α-hydrogen to form an enolate, which then undergoes an SN2 reaction with a primary or methyl alkyl halide.
This method is applicable to ketones, esters, and nitriles.
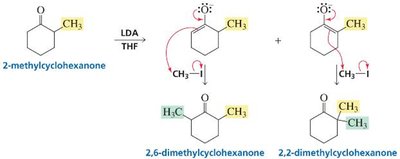
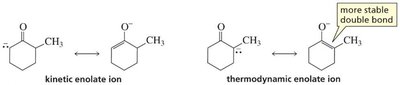
Regioselectivity: Kinetic vs. Thermodynamic Enolate
Asymmetrical ketones with α-hydrogens on both sides can yield two products.
The kinetic enolate forms faster (less substituted, more accessible α-hydrogen), while the thermodynamic enolate is more stable (more substituted double bond).
Strong, non-nucleophilic bases (e.g., LDA) favor kinetic enolate; weaker bases (e.g., HO−) favor thermodynamic enolate.
Aldol Addition and Condensation
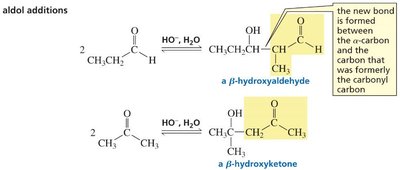
Aldol Addition: Formation of β-Hydroxyaldehydes and β-Hydroxyketones
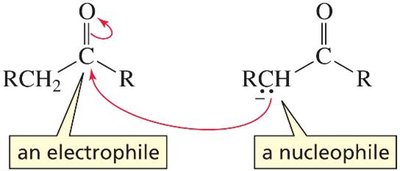
An aldol addition involves two molecules of an aldehyde or ketone. One acts as a nucleophile (enolate ion), the other as an electrophile (carbonyl carbon). The product is a β-hydroxyaldehyde or β-hydroxyketone, depending on the starting material.
The reaction forms a new C–C bond between the α-carbon of one molecule and the carbonyl carbon of the other.
Retro-Aldol Reaction
Aldol reactions are reversible. Heating the β-hydroxyaldehyde or β-hydroxyketone with base and water regenerates the original carbonyl compounds.
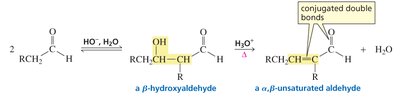
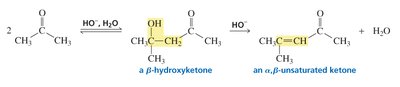
Aldol Condensation: Dehydration to α,β-Unsaturated Carbonyls
Upon heating with base, the aldol addition product loses water to form an α,β-unsaturated aldehyde or ketone. The resulting conjugated system is stabilized by resonance, making the dehydration favorable.
Crossed Aldol Addition
When two different carbonyl compounds are used, four possible products can form due to the formation of two different enolate ions and two possible electrophiles. Synthetic utility is improved by using conditions that favor the formation of a single product (e.g., using a compound with no α-hydrogens).
Claisen Condensation
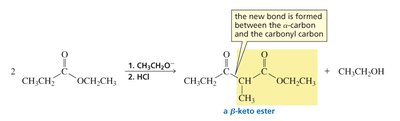
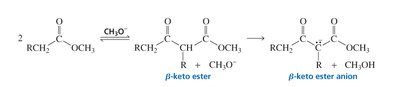
Formation of β-Keto Esters
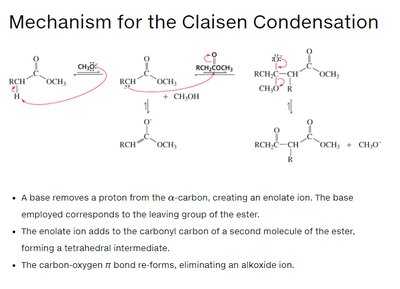
Claisen condensation is the reaction of two esters (or one ester and one ketone) to form a β-keto ester. Like the aldol addition, one molecule acts as a nucleophile (enolate ion), the other as an electrophile (carbonyl carbon). The reaction proceeds via nucleophilic acyl substitution, eliminating an alkoxide ion.
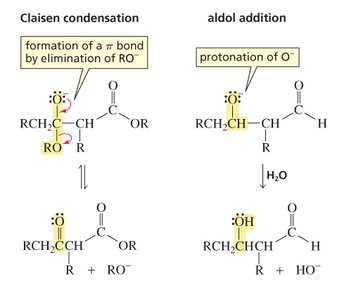
Mechanistic Comparison: Claisen vs. Aldol
In Claisen condensation, the negatively charged oxygen reforms the C=O bond, eliminating the -OR group (nucleophilic acyl substitution).
In aldol addition, the negatively charged oxygen is simply protonated (nucleophilic addition).
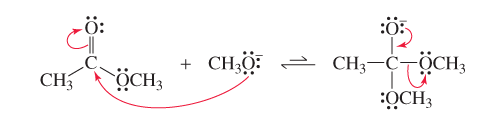
Requirements and Driving Force
Claisen condensation requires an ester with two α-hydrogens for successful reaction.
The reaction is reversible but can be driven to completion by deprotonation of the β-keto ester product, which is stabilized by resonance between two carbonyl groups.
Crossed Claisen Condensation
A crossed Claisen condensation involves two different esters. It is synthetically useful only if one ester lacks α-hydrogens (cannot form an enolate), and the other is added slowly to minimize side products.
Additional info: The study of α-carbon reactivity is foundational for understanding carbon–carbon bond formation in organic synthesis, including the construction of complex molecules in pharmaceuticals and natural products.
