 Back
BackProtein Analysis: Techniques and Applications in Biochemistry
Study Guide - Smart Notes
 Tailored notes based on your materials, expanded with key definitions, examples, and context.
Tailored notes based on your materials, expanded with key definitions, examples, and context.
Protein Analysis
Overview
Protein analysis is essential in biochemistry for determining the concentration, structure, sequence, and identity of proteins. Various analytical techniques are used to characterize proteins, each with specific applications, advantages, and limitations.
Protein Concentration Determination
Methods for Protein Quantification
Several methods are used to determine protein concentration in a sample. These methods are based on different principles and are chosen based on sample type, required sensitivity, and compatibility with other reagents.
UV Absorbance at 280 nm (A280 Assay): Measures absorbance of aromatic amino acids (Tyr, Trp, Phe) at 280 nm. Quick and reagent-free but can be affected by nucleic acids and other UV-absorbing compounds.
Bradford Assay: Uses Coomassie Brilliant Blue dye, which binds to proteins and shifts absorbance to 595 nm. Fast and sensitive, but sensitivity varies with protein type and can be affected by detergents like SDS.
Bicinchoninic Acid (BCA) Assay: Based on Cu2+ reduction by peptide bonds, detected with BCA reagent at 562 nm. Compatible with detergents but sensitive to reducing agents and chelators.
Lowry Assay: Involves Cu2+ reduction followed by Folin–Ciocalteu reaction at 750 nm. Highly sensitive but time-consuming and affected by detergents/reducing agents.
Biuret Assay: Cu2+ reacts with peptide bonds at 540 nm. Simple and reliable but has low sensitivity.
Kjeldahl Method: Measures nitrogen content via acid digestion and titration. Highly accurate but labor-intensive and uses hazardous chemicals.
Dumas Method: High-temperature combustion measures nitrogen content. Fast and accurate but requires specialized equipment.
Polyacrylamide Gel Electrophoresis (PAGE)
Principle and Gel Formation
Polyacrylamide gel electrophoresis separates proteins based on their size and charge. The gel is formed by polymerizing acrylamide with a crosslinker, N,N'-methylenebisacrylamide. Adjusting the concentration of these components changes the pore size of the gel, allowing for the separation of proteins of different sizes.
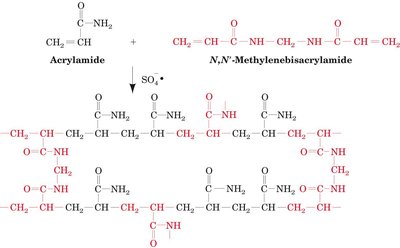
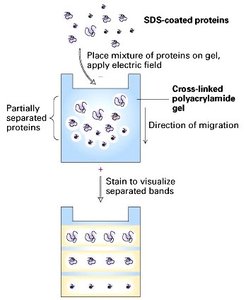
SDS-PAGE (Sodium Dodecyl Sulfate PAGE)
SDS-PAGE is a denaturing electrophoresis technique that separates proteins primarily by mass. SDS, a detergent, binds to proteins, disrupting their secondary and tertiary structures and conferring a uniform negative charge. Reducing agents (e.g., β-mercaptoethanol, DTT) are often used to break disulfide bonds, ensuring proteins are fully denatured and linearized. As a result, protein mobility in the gel depends only on size.
Denaturing: Proteins are unfolded and coated with SDS.
Separation by Mass: Proteins migrate through the gel based on size; smaller proteins move faster.
Resolution: Can resolve complex mixtures of proteins.
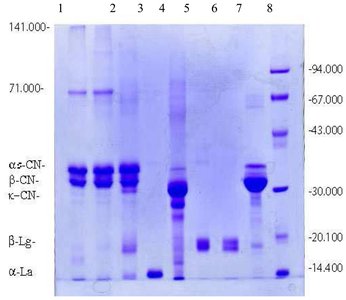
Example: SDS-PAGE Gel
SDS-PAGE gels are visualized by staining, revealing bands corresponding to proteins of different molecular weights. These gels are used to assess protein purity and estimate molecular weight.
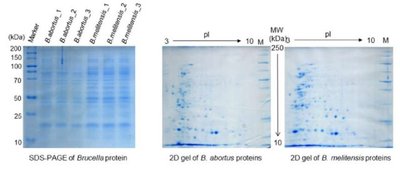
Two-Dimensional Electrophoresis (2D Electrophoresis)
Principle and Application
2D electrophoresis separates proteins in two dimensions: first by isoelectric point (pI) and then by molecular weight. This technique provides higher resolution than 1D electrophoresis, allowing for the separation of proteins with similar sizes but different pI values.
First Dimension: Isoelectric focusing separates proteins based on their pI.
Second Dimension: SDS-PAGE separates proteins by size.
Application: Used for complex protein mixtures, such as in proteomics.
Protein Sequence Determination
Edman Degradation
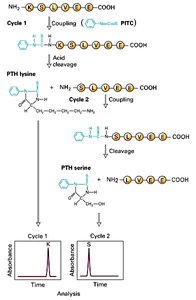
Edman degradation is a method for sequencing amino acids in a peptide. The N-terminal amino acid is labeled and cleaved without disrupting the peptide bonds of other amino acids, allowing sequential identification of up to 40–60 residues.
Labeling: Phenylisothiocyanate (PITC) reacts with the N-terminal amino acid.
Cleavage: The labeled amino acid is cleaved and identified.
Repetition: The process is repeated for the next residue.
Peptide Mapping
Enzymatic Proteolysis and Peptide Maps
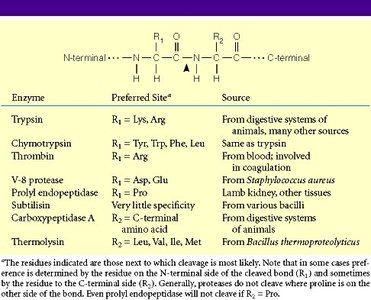
Peptide mapping involves enzymatic digestion of proteins (e.g., with trypsin) to produce a unique set of peptides. These peptides are separated and analyzed, providing a "fingerprint" for protein identification.
Specificity: Proteases cleave at specific amino acid residues, generating predictable fragments.
Analysis: Peptide fragments are separated by gel electrophoresis or chromatography.
Application: Used to confirm protein identity and detect modifications.
Mass Spectrometry in Protein Analysis
Principle of Mass Spectrometry
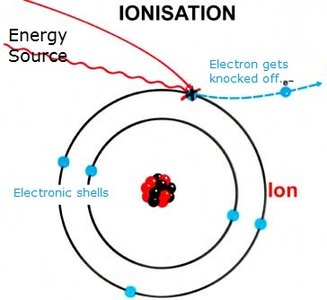
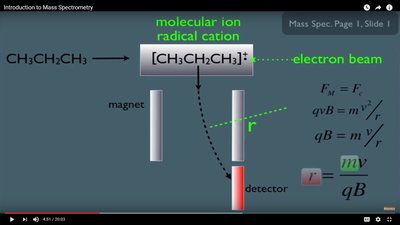
Mass spectrometry (MS) identifies atoms and molecules based on their mass-to-charge ratio (m/z). It is a powerful tool for protein identification, sequencing, and characterization.
Ionization: Converts molecules into charged ions.
Deflection/Detection: Ions are separated by m/z and detected.
Fragmentation: Fragmentation patterns provide sequence information.
Ionization Techniques
Electrospray Ionization (ESI): Produces ions from liquid samples, suitable for coupling with liquid chromatography (LC-MS).
Matrix-Assisted Laser Desorption Ionization (MALDI): Uses a laser to ionize proteins embedded in a matrix, preserving post-translational modifications.
Deflection and Detection
In MS, ions are deflected by magnetic or electric fields. The degree of deflection depends on the mass-to-charge ratio, allowing separation and detection of different ions.
Proteomics: Top-Down and Bottom-Up Approaches
Strategies for Protein Identification
Proteomics uses mass spectrometry to identify and characterize proteins in complex mixtures. Two main strategies are employed:
Top-Down Proteomics: Analyzes intact proteins, often using MALDI. Provides information on post-translational modifications and protein isoforms.
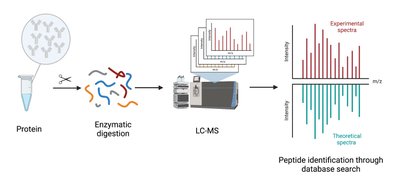
Bottom-Up Proteomics: Proteins are digested into peptides (e.g., by trypsin), then analyzed by LC-MS/MS. Allows high-throughput identification and quantification.

LC-MS Workflow
In bottom-up proteomics, proteins are enzymatically digested, separated by liquid chromatography, and analyzed by mass spectrometry. Peptide mass fingerprints are matched to databases for identification.
Mass Spectrometry for Protein Sequencing
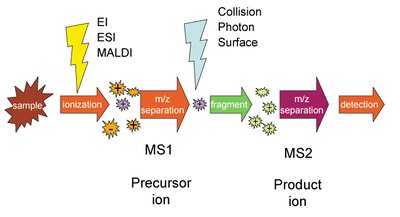
MS/MS (Tandem Mass Spectrometry)
MS/MS involves two stages of mass analysis. Peptides are first separated by m/z, then selected ions are fragmented, and the resulting fragments are analyzed to deduce the amino acid sequence.
Fragmentation: Collision-induced dissociation (CID) breaks peptides into smaller fragments.
Sequence Reconstruction: Differences in fragment masses correspond to specific amino acids, allowing sequence determination.
Proteomics and Genomic Search Space
Database Search for Protein Identification
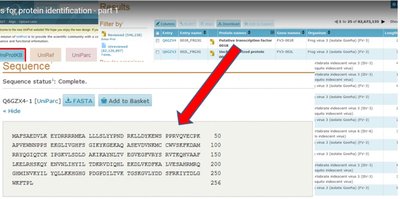
Mass spectrometry data are compared to theoretical spectra generated from genomic and proteomic databases. This reduces the search space and increases confidence in protein identification.
Peptide Mass Fingerprint (PMF): Unique peptide masses identify proteins.
Database Matching: Experimental spectra are matched to predicted spectra from known protein sequences.
Summary Table: Protein Quantification Methods
Method | Principle | Wavelength (nm) | Advantages | Limitations | Reagents Used |
|---|---|---|---|---|---|
A280 Assay | Absorbance of aromatic amino acids | 280 | Quick, reagent-free | Interference from nucleic acids | None |
Bradford Assay | Dye binding (Coomassie Brilliant Blue) | 595 | Fast, sensitive | Protein-to-protein variation, SDS interference | Coomassie dye, acidic buffer |
BCA Assay | Cu2+ reduction, BCA detection | 562 | Detergent compatible | Reducing agent interference | BCA, CuSO4, buffer |
Lowry Assay | Cu2+ reduction, Folin–Ciocalteu | 750 | High sensitivity | Time-consuming, interference | Copper sulfate, Folin–Ciocalteu |
Biuret Assay | Cu2+ reacts with peptide bonds | 540 | Simple, reliable | Low sensitivity | Biuret reagent |
Kjeldahl Method | Nitrogen content by acid digestion | N/A | Highly accurate | Labor-intensive, hazardous | H2SO4, NaOH, H3BO3, catalyst |
Dumas Method | Nitrogen by combustion | N/A | Fast, accurate | Specialized equipment | High-temp combustion gases |
