 Back
BackProtein Degradation and Amino Acid Catabolism: The Ubiquitin–Proteasome Pathway and Nitrogen Metabolism
Study Guide - Smart Notes
 Tailored notes based on your materials, expanded with key definitions, examples, and context.
Tailored notes based on your materials, expanded with key definitions, examples, and context.
Protein Degradation: The Ubiquitin–Proteasome Pathway
Why Do Cells Degrade Proteins?
Protein degradation is essential for maintaining cellular homeostasis and regulating protein levels. Cells degrade proteins to remove damaged or misfolded proteins, control the cell cycle and signaling, and maintain a balanced, functional proteome. This process, known as proteostasis, ensures that proteins are synthesized, folded, trafficked, and degraded appropriately to prevent the accumulation of toxic or aggregated proteins.
Proteostasis: Dynamic regulation of the protein population within and outside the cell.
Functions: Removal of damaged proteins, regulation of protein levels, and control of cellular processes.
Overview of the Ubiquitin–Proteasome System (UPS)
The Ubiquitin–Proteasome System (UPS) is an ATP-dependent pathway that targets specific intracellular proteins for degradation. Ubiquitin acts as a molecular tag, and the proteasome is the proteolytic complex that degrades tagged proteins.
ATP-dependent: Requires energy for ubiquitin activation and protein unfolding.
Specificity: Targets proteins with specific degradation signals (degrons).
Proteasome: Multisubunit complex responsible for proteolysis.
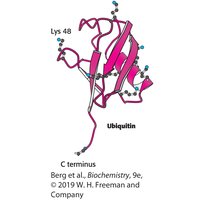
What is Ubiquitin?
Ubiquitin is a small, 76-amino acid protein that is highly conserved in eukaryotes. It is covalently attached to lysine residues of substrate proteins, often forming polyubiquitin chains that signal for degradation.
Structure: Compact, globular protein with a C-terminal glycine and several lysine residues (notably Lys48).
Function: Marks proteins for degradation via the proteasome.
Mechanism of Ubiquitin Attachment
The attachment of ubiquitin to substrate proteins is a multi-step enzymatic process involving three main enzymes: E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase). The process is ATP-dependent and results in the formation of an isopeptide bond between the C-terminal glycine of ubiquitin and the ε-amino group of a lysine residue on the substrate.
E1: Activates ubiquitin using ATP.
E2: Receives ubiquitin from E1 and prepares it for transfer.
E3: Recognizes the substrate and facilitates the transfer of ubiquitin from E2 to the target protein.
Isopeptide bond: Covalent linkage between ubiquitin and substrate lysine.
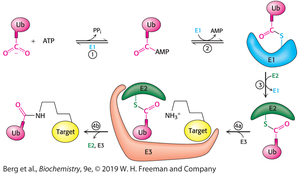
The Ubiquitination Enzyme Cascade
The ubiquitination process is highly specific due to the large number of E3 ligases, each recognizing different substrates. Polyubiquitin chains, especially those linked through Lys48, target proteins for proteasomal degradation.
E1: Ubiquitin-activating enzyme (few types).
E2: Ubiquitin-conjugating enzyme (dozens of types).
E3: Ubiquitin ligase (hundreds of types, substrate specificity).
Polyubiquitination: Multiple ubiquitin molecules attached in a chain.
Ubiquitin Tags Proteins for Destruction
Specific amino acid sequences, called degrons, determine the half-life of proteins. The N-terminal residue, cyclin destruction boxes, and PEST sequences are common degrons. Proteins may require modification to expose degrons for recognition by E3 ligases.
Degrons: Sequence motifs that signal for degradation.
N-end rule: The identity of the N-terminal amino acid affects protein stability.
PEST sequences: Regions rich in Pro, Glu, Ser, and Thr, associated with rapid degradation.
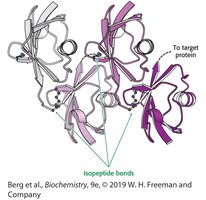
Structure of Tetraubiquitin
Polyubiquitin chains are formed by linking the C-terminal glycine of one ubiquitin to the lysine residue (often Lys48) of another. This structure is recognized by the proteasome for degradation.
Importance of E3 Proteins to Normal Cell Function
E3 ubiquitin ligases are crucial for cellular regulation. Defects in E3 ligases can lead to diseases such as Parkinson's disease, Angelman syndrome, autism, and cancer due to inappropriate protein turnover.
Defective E3: Accumulation of proteins, leading to aggregation diseases.
Overactive E3: Excessive degradation of essential proteins, such as tumor suppressors.
The Proteasome Digests the Ubiquitin-tagged Proteins
The 26S proteasome is a large proteolytic complex composed of a 20S catalytic core and two 19S regulatory units. The regulatory units recognize polyubiquitin chains, remove ubiquitin, unfold the substrate, and translocate it into the core for degradation.
20S core: Catalytic chamber where proteolysis occurs.
19S regulatory unit: Recognizes ubiquitinated proteins, removes ubiquitin, and unfolds substrates.
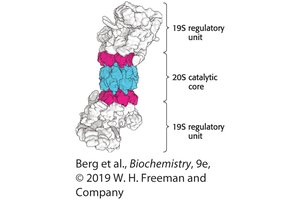
20S Proteasome Structure
The 20S proteasome consists of 28 subunits arranged in four rings (two α-type and two β-type). The β-type subunits contain the protease active sites. The structure allows for selective entry and degradation of proteins.
Subunit arrangement: Four rings of seven subunits each.
Active sites: Located in β-type subunits.
Biological and Medical Relevance
Protein degradation regulates many biological processes. Inhibitors of the proteasome, such as bortezomib (Velcade), are used in cancer therapy. Other inhibitors target bacterial proteasomes for infectious disease treatment.
Bortezomib: Used to treat multiple myeloma.
HT1171: Inhibits the proteasome of M. tuberculosis.
Amino Acid Degradation and Nitrogen Metabolism
The First Step in Amino Acid Degradation: Removal of Nitrogen
Amino acid catabolism begins with the removal of the α-amino group, which is funneled to glutamate via aminotransferases. Glutamate is then deaminated to release ammonium ions (NH4+).
Aminotransferases: Transfer amino groups to α-ketoglutarate, forming glutamate.
Glutamate dehydrogenase: Releases NH4+ from glutamate, using NAD+ or NADP+ as cofactors.
Blood Levels of Aminotransferases as Diagnostic Markers
Elevated levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in the blood indicate liver damage, as these enzymes leak from damaged hepatocytes.
Clinical relevance: Used to diagnose liver diseases such as hepatitis and alcohol-induced liver injury.
Direct Deamination of Serine and Threonine
Serine and threonine can be directly deaminated by serine dehydratase and threonine dehydratase, respectively. Dehydration precedes deamination in these reactions.
Nitrogen Transport to the Liver
Peripheral tissues transport nitrogen to the liver via the glucose–alanine cycle and as glutamine. Muscle uses branched-chain amino acids as fuel, and the resulting nitrogen is transported as alanine or glutamine.
Glucose–alanine cycle: Transfers nitrogen from muscle to liver.
Glutamine synthetase: Converts glutamate to glutamine for safe transport.
The Urea Cycle
Excess ammonium ions are converted to urea in the liver via the urea cycle. This cycle involves several steps and enzymes, beginning with the formation of carbamoyl phosphate and ending with the production of urea and regeneration of ornithine.
Key enzymes: Carbamoyl phosphate synthetase I, ornithine transcarbamoylase, argininosuccinate synthetase, argininosuccinase, arginase.
Regulation: N-acetylglutamate activates carbamoyl phosphate synthetase I.
Stoichiometry: Fumarate produced can enter gluconeogenesis.
Evolutionary Relationships and Inherited Defects
Several urea cycle enzymes are evolutionarily related to enzymes in nucleotide biosynthesis. Inherited defects in the urea cycle cause hyperammonemia and can lead to neurological damage. Treatments include dietary supplementation and alternative pathways for nitrogen excretion.
Examples: Argininosuccinate deficiency (treated with arginine), carbamoyl phosphate synthetase deficiency (treated with benzoate or phenylacetate).
Alternative Nitrogen Excretion Strategies
Different organisms excrete nitrogen in various forms: urea (ureotelic), ammonium (ammoniotelic), or uric acid (uricotelic).
Fates of Amino Acid Carbon Skeletons
The carbon skeletons of amino acids are metabolized to seven major intermediates: pyruvate, acetyl CoA, acetoacetyl CoA, α-ketoglutarate, succinyl CoA, fumarate, and oxaloacetate. Amino acids are classified as ketogenic, glucogenic, or both based on their metabolic fate.
Ketogenic: Leucine and lysine (form fats, not glucose).
Glucogenic: Most other amino acids (can be used for glucose synthesis).
Entry Points for Amino Acid Degradation
Pyruvate: Alanine, serine, cysteine, glycine, threonine, tryptophan.
Oxaloacetate: Aspartate, asparagine.
α-Ketoglutarate: Glutamate, glutamine, proline, arginine, histidine.
Succinyl CoA: Methionine, isoleucine, valine, threonine.
Acetyl CoA/Acetoacetyl CoA: Leucine, lysine, phenylalanine, tyrosine, tryptophan.
Degradation of Branched-Chain and Aromatic Amino Acids
Branched-chain amino acids (leucine, isoleucine, valine) are degraded via pathways similar to fatty acid oxidation. Aromatic amino acids (phenylalanine, tyrosine, tryptophan) require oxygenases for ring cleavage and further metabolism.
Phenylalanine hydroxylase: Converts phenylalanine to tyrosine (requires tetrahydrobiopterin).
Dioxygenases: Cleave aromatic rings in tryptophan degradation.
Inborn Errors of Amino Acid Metabolism
Genetic defects in amino acid degradation pathways can lead to metabolic diseases:
Alcaptonuria: Deficiency of homogentisate oxidase (dark urine).
Maple syrup urine disease: Deficiency of branched-chain α-ketoacid dehydrogenase.
Phenylketonuria (PKU): Deficiency of phenylalanine hydroxylase (neurological impairment).
Methylmalonic acidemia: Accumulation of methylmalonic acid due to defective conversion of propionyl CoA to succinyl CoA.
Clinical and Physiological Relevance
Protein and amino acid metabolism is crucial for energy production, especially during fasting or intense activity (e.g., migratory birds). Disruptions in these pathways can have severe physiological and neurological consequences.
